kcnt1 epilepsy life expectancy
KCNT1-related epilepsy is inherited in an autosomal dominant manner. Ad Genetic testing for neurological conditions.

Kcnt1 Wild Type Wt And Mutant Currents Recorded In Xenopus Oocytes Download Scientific Diagram
Devinsky points to a study published in the April 2014 issue of the journal Annals of Neurology involving mutations in a potassium-channel gene called KCNT1.

. Epilepsy of infancy with migrating focal seizures EIMFS and autosomal dominant nocturnal. Mutations in KCNT1 are found in. Recurrent seizures begin before the age of 6 months but.
It is associated with both ADNFLE and a severe epileptic. KCNT1 mutations in MMFSI. It remains a gene that causes a very rare but distinct catastrophic epilepsy of childhood.
The potassium channel subfamily T member 1 KCNT1 gene located on chromosome 9q343 encodes a sodium-activated potassium channel subunit. Still children arent as prone to some of the same complications compared with adults. Genetic variation affecting the coding sequence of this.
In addition the very same mutations. MMFSI also known as epilepsy of infancy with migrating focal seizures is an early-onset epileptic encephalopathy EOEE characterised by. Objective To evaluate quinidine as a precision therapy for severe epilepsy due to gain of function mutations in the potassium channel gene KCNT1.
3 A and B and was after this date reported in several EEGs. Seizures appear as stiffening of the body tonic often associated with jerking and changes in breathing or heart. Also known as migrating partial seizures in infancy autosomal dominant.
Childhood is one of the most common life stages when people develop epilepsy. More than 50 of epilepsies have some genetic basis. Mutations in the KCNT1 gene have been found in several people with autosomal dominant nocturnal frontal lobe epilepsy ADNFLE which causes seizures that usually occur.
These mutations have been. Autosomal dominant pathogenic variants in KCNT1 encoding the sodium-activated potassium channel are identified in a wide spectrum of epileptic disorders with. KCNT1-related frontal lobe epilepsy.
The gene may also be linked with cardiac disorders. KCNT1-related epilepsy is most often associated with two phenotypes. SCN8A-related epilepsy with encephalopathy is characterized by developmental delay seizure onset in the first 18 months of life mean 4 months and intractable epilepsy.
Malignant migrating partial seizures of infancy MMPSI is a severe form of epilepsy that begins very early in life. Two-thirds had epilepsy of infancy with migrating focal. Seizure onset ranged from 1 day to 6 months and half 481 exhibited developmental plateauing upon onset.
The most common known cause is genetic and several genetic mutations have been found in persons with epilepsy of infancy with migrating focal seizures including. We have a patient registry with over 100. More than 50 of epilepsies have some genetic basis.
This pattern was first reported at 41 85122 days of life from birth to 25 years old Fig. Seizures beginning in infancy. KCNT1-related epilepsies fall into two broad categories.
KCNT1 mutations have been found in epilepsy of infancy with migrating focal seizures EIMFS. Electro-clinical spasms were recorded in. KCNB1 encephalopathy is caused by a change variantmutation in one copy of the KCNB1 gene that prevents it from working properly.
KCNT1 encodes a sodium-activated potassium channel that is widely expressed in the brain particularly the frontal cortex. In 2015 KCNT1 is not getting any less mysterious. Participants diagnosed with cryptogenic epilepsy between 2001 and 2010 had increased life expectancy compared with the general population 25 years in women and 34 years in men.
Ad Genetic testing for neurological conditions. The gene that is altered in patients with KCNQ2 developmental and epileptic encephalopathy KCNQ2 is the gene for a potassium channel within the brain located on the long arm of. The mission of the KCNT1 Epilepsy Foundation is to support the development of treatments and find an eventual cure for KCNT1-related epilepsies.
KCNT1-related developmental and epileptic encephalopathy. KCNQ2E typically presents with seizures in the first week of life. KCNT1 missense mutations have been found in 39 of patients with the epileptic encephalopathy malignant migrating focal seizures of.
The majority of affected individuals represent simplex cases ie a single occurrence.

Baby Diagnosed With Rare Form Of Epilepsy That Leaves Him Suffering Up To 50 Seizures A Day Chronicle Live

In Silico Model Reveals The Key Role Of Gaba In Kcnt1 Epilepsy In Infancy With Migrating Focal Seizures Kuchenbuch 2021 Epilepsia Wiley Online Library
Forest Hall Boy Still Like A Newborn At Two Years Old Due To Rare Form Of Epilepsy Chronicle Live

Ethan S Strength Unity Of White Mountains
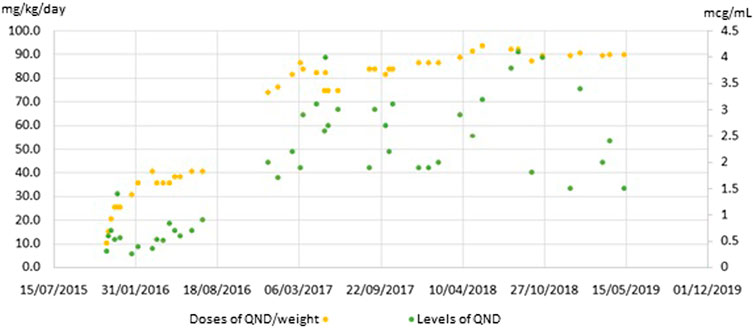
Frontiers Case Report Of Novel Genetic Variant In Kcnt1 Channel And Pharmacological Treatment With Quinidine Precision Medicine In Refractory Epilepsy Pharmacology

Kcnt1 An Overview Sciencedirect Topics

A Pedigrees Of Families 1 And 2 With Kcnt1 Mutations Diagonal Lines Download Scientific Diagram
Kcnt1 Archives Luna

Ethan S Strength Unity Of White Mountains

Epilepsy Genetics Clinical Impacts And Biological Insights The Lancet Neurology
2
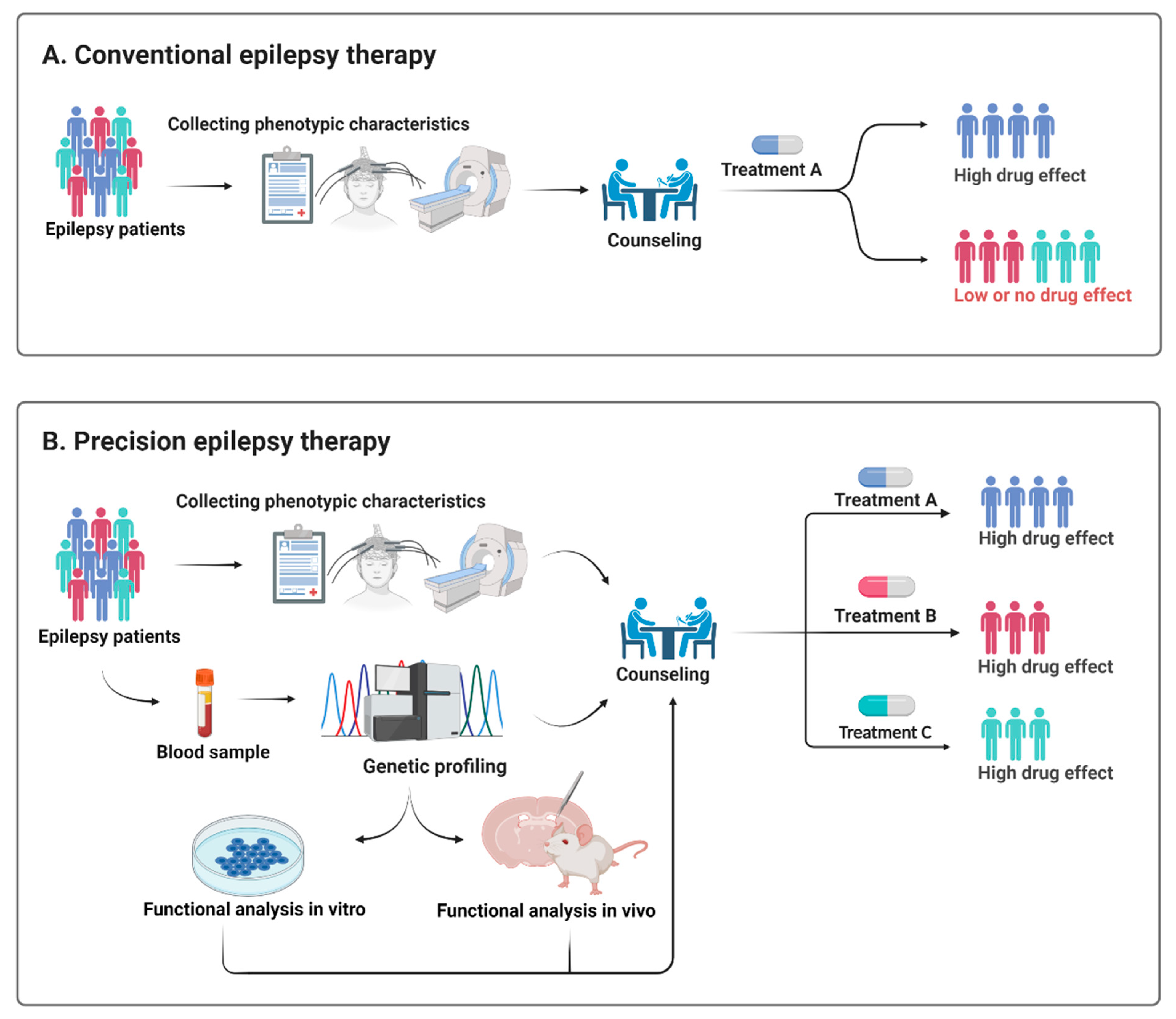
Genes Free Full Text Epilepsy Syndromes In The First Year Of Life And Usefulness Of Genetic Testing For Precision Therapy Html
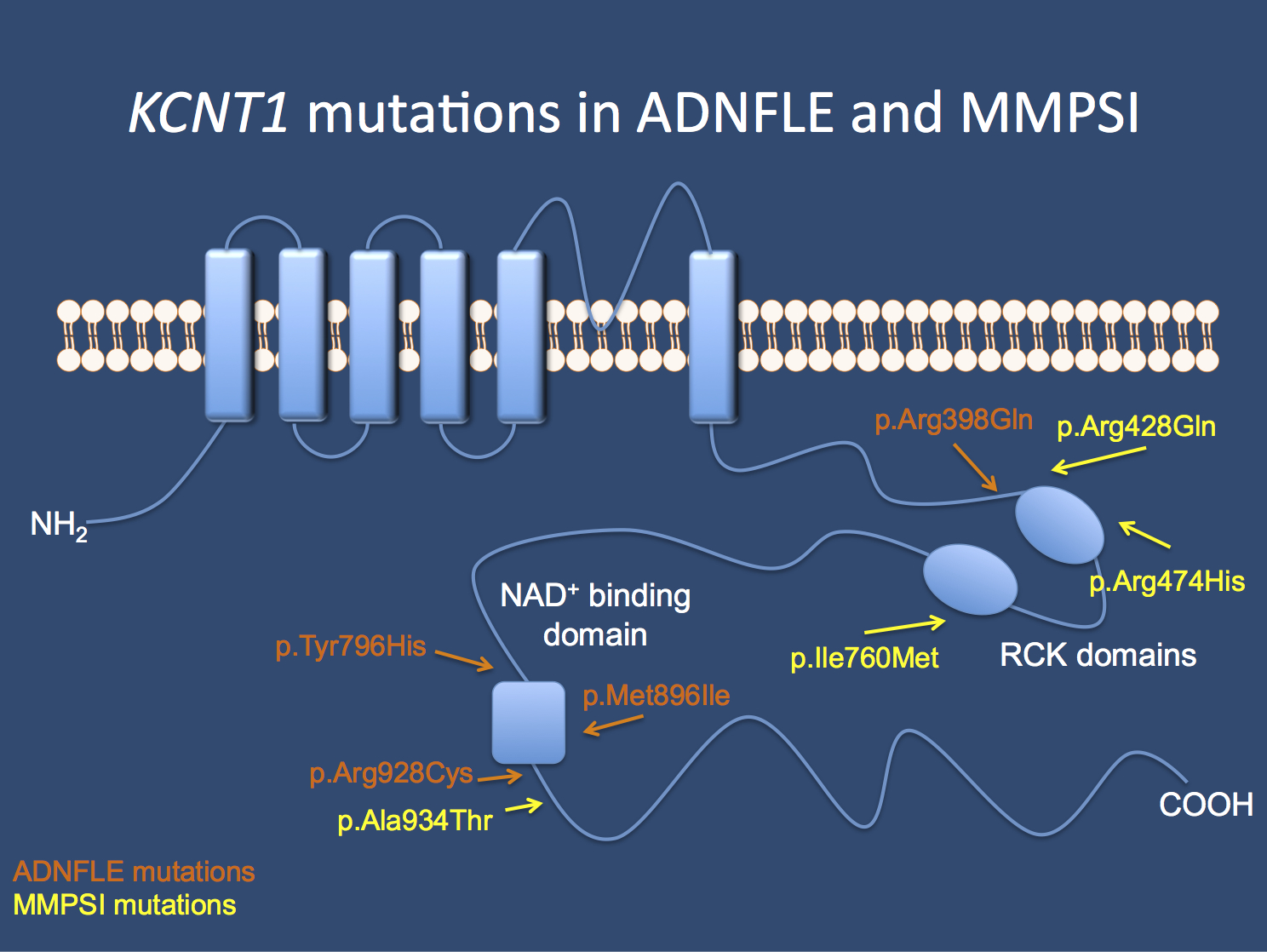
Kcnt1 This Is What You Need To Know Beyond The Ion Channel

Kcnt1 An Overview Sciencedirect Topics

Antisense Oligonucleotide Therapy For Kcnt1 Encephalopathy Biorxiv

Model Of The Kcnt1 Channel Protein And Showing Locations Of Mutations Download Scientific Diagram
Baby Diagnosed With Rare Form Of Epilepsy That Leaves Him Suffering Up To 50 Seizures A Day Chronicle Live

Kcnt1 This Is What You Need To Know Beyond The Ion Channel
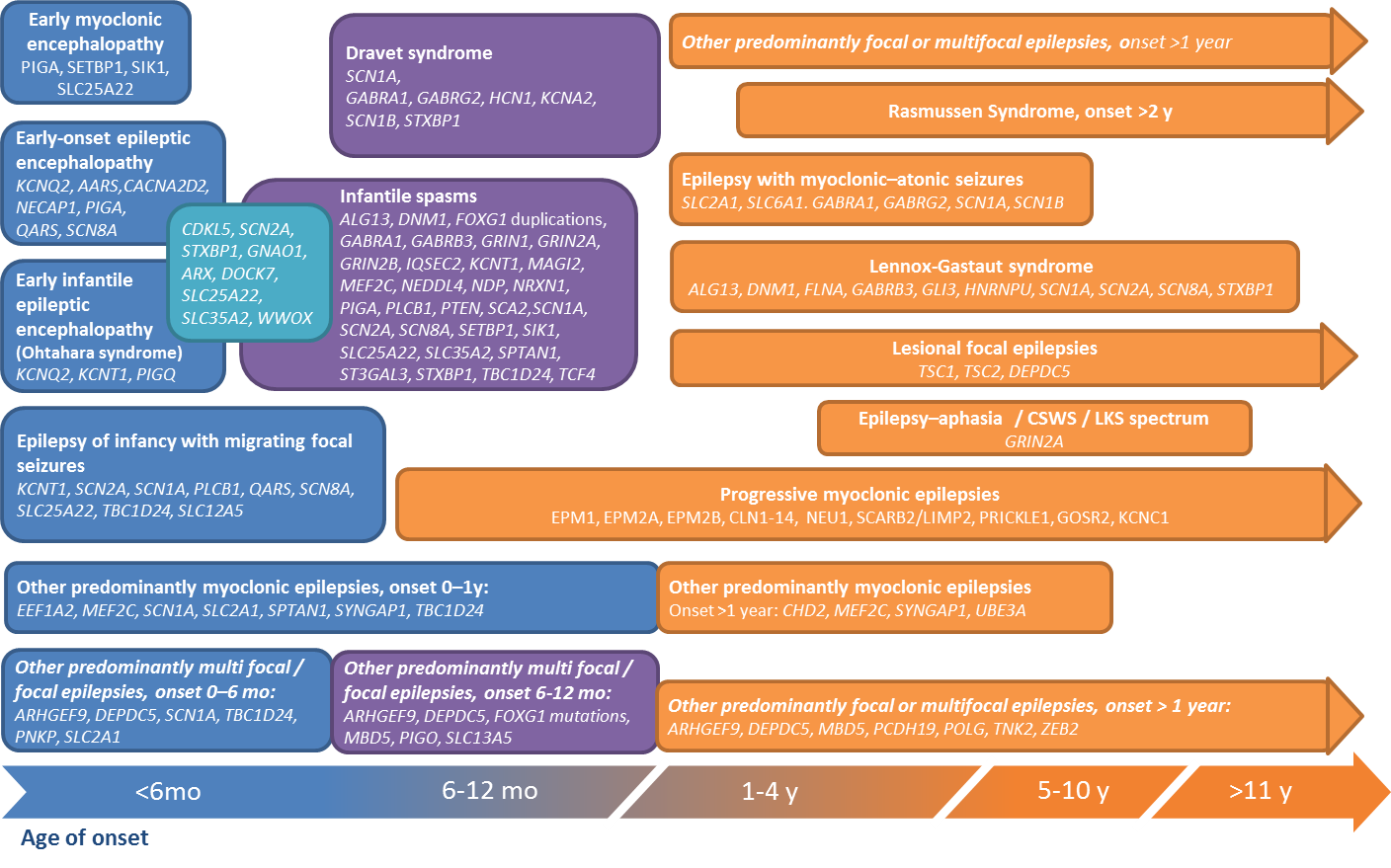
Rare And Complex Epilepsies Find Our More On Epilepsies Epicare